Epigenetic Mechanisms in Post-Traumatic Stress Disorder
Andrea Burri, PhD; Andreas Küffer, MSc; Andreas Maercker, MD, PhD; Department of Psychology, University of Zürich, Switzerland; University Research Priority Program “Dynamics of Healthy Aging,” University of Zürich, Switzerland
January 16, 2013
Evidence from quantitative and molecular genetic studies have repeatedly demonstrated the involvement of genes in post-traumatic stress disorder (PTSD), but also highlighted the importance of environmental factors in disease development and/or resilience. The emerging field of epigenetics is particularly attractive because of its ability to account for individual differences in response to trauma based on environmental exposures that alter gene function. As such, investigation of specific epigenetic markers may provide quantifiable measures of lifetime environmental stress or heritable predisposition to PTSD and can thus offer a biological framework for the interactive causes underlying PTSD. In this mini-review we will discuss the relevance of epigenetic research to PTSD and provide a summary of recent developments revealing epigenetic alterations in response to adversities, trauma and PTSD.
Epigenetics Explained in Brief
Coined by Waddington, the term “epigenetics” was originally used to describe how genes interact with the cellular environment to produce a phenotype (Waddington, 1942). The term is now applied more narrowly, referring to the reversible regulation of various genomic functions, occurring independently of DNA sequence, mediated principally through changes in DNA methylation and chromatin structure. In other words, epigenetics defines cellular modifications that can be heritable but appear largely unrelated to DNA sequence changes, and that can be modified by environmental stimuli (Holliday, 1994, Russo et al.,1996). Such modifications are stable and long lasting and can in some cases be transmitted inter-generationally (Meaney & Szyf, 2005). At present, epigenetic mechanisms typically comprise DNA methylation and histone modifications. DNA methylation is strongly linked to a number of genomic functions, including the regulation of gene expression, with many genes demonstrating an inverse correlation between the degree of methylation and the level of expression (Jaenisch & Bird, 2003). Genome-wide DNA methylation variation is a new way of looking at complex diseases such as PTSD because it responds to the environment and governs gene expression - thus potentially mediating a path from environmental effects to gene expression to disease.
Why Investigating Epigenetics in PTSD?
Apart from the evidence supporting a genetic predisposition to PTSD, the importance of environmental stressors in disease development has been supported by the findings from genetic research, highlighting the crucial existence of gene-environment interactions (GxE) in the pathogenesis of the condition (see Illustration; Binder et al., 2008; Bradley et al., 2008). A G×E interaction occurs when the effect of a specific genotype on risk of a disorder differs by the presence or absence of an environmental pathogen (Koenen et al., 2008). Although results of such studies may explain some of the individual differences in trauma exposure and vulnerability to PTSD, the epigenetic model differs from that associated with traditional G×E interactions as it explains how environmental exposure alters the function of the gene which might then govern an individual’s response to a subsequent traumatic event (Meaney & Szyf, 2005). Epigenetic mechanisms therefore offer an additional explanation for the impact of personal history on vulnerability, the cumulative effects of repeated exposures, and intergenerational influences that are reflective of experience (Yehuda & Bierer, 2009).
Epigenetic Alterations in PTSD: Evidence from Animal and Human Research
Whilst epigenetic research in human is still in its early stages, there is robust evidence for an epigenetic contribution to the development of PTSD-like symptoms in rodents. Several studies have repeatedly demonstrated the impact of maternal behavior on hypothalamic corticotrophin-releasing hormone expression and the development of individual differences in hypothalamic-pituitary-adrenal (HPA) stress responses (e.g. Szyf et al., 2005; Meaney, 2001; Higley et al., 1991).
In a study conducted on rat mothers and their offspring, Szyf and colleagues found evidence for how epigenomic alterations can be induced through a behavioral mode of programming and how they are dynamic and potentially reversible. In their study, increased levels of pup grooming by the mothers lead to changes in DNA methylation at the glucocorticoid receptor gene promoter in the hippocampus of the offspring. Supporting these findings, Weaver et al. (2006) also unveiled epigenetic changes occurring in the glucocorticoid receptor gene in response to maternal care. Another recent study from Roth et al. (2011) found significant epigenetic alterations of the brain-derived neurotrophic factor (BDNF) gene in rats exposed to psychosocial stress, providing evidence for a link between traumatic stress and CNS gene methylation.
The authors concluded by speculating that such altered hippocampal BDNF DNA methylation might be a cellular mechanism underlying the persistent cognitive deficits which are prominent features of the pathophysiology of PTSD. In yet another mouse study conducted by Renthal et al. (2007), histone deacetylase 5 (HDAC5) emerged as an epigenetic regulator of chronic stress and its related long-term pathophysiologic status with implications for the understanding of biological changes occurring during prolonged exposure to stress.
Epigenetic research in humans has also come up with promising findings. Probably the most noteworthy study was conducted by Klengel and colleagues on a predominantly African-American, highly and repeatedly traumatized patient cohort of n > 4000 males and females (Klengel et al., 2012). The study provides evidence for an epigenetic mechanism that mediates the combined effects of childhood adversity and a genetic polymorphism on the risk of developing stress-related dis¬orders by reporting childhood trauma–dependent DNA demethylation in functional glucocorticoid response elements of the FK506 binding protein 5 (FKBP5) gene. As such the authors provide insight into the molecular mechanisms and consequences of gene × environment interaction, facilitat¬ing a better understanding of the pathophysiology of stress-related psychiatric disorders.
In another recent study by Ressler and colleagues (2011) methylation of the first CpG-rich island of the ADCYAP1R1 gene was significantly associated with total PTSD symptomology and PTSD diagnosis but not with other disorders (e.g. depression). ADCYAP1R1 encodes for a selective peptide receptor in hypothalamic and limbic structures and its function has been demonstrated to be integrally involved in stress and anxiety like behavior in rodents. In an impressive study using human postmortem brains obtained from suicide victims with a history of childhood abuse and healthy controls, McGowan et al. (2009) further found decreased levels of glucocorticoid receptor mRNA and increased CpG methylation levels in suicide victims with a history of childhood abuse, as compared with suicide victims without a history of abuse. Examining the link between methylation of the serotonin transporter coding gene SLC6A4 and PTSD whilst controlling for the genotype, Koenen et al. (2011) demonstrated how SLC6A4 methylation influences the relationship between number of traumatic events and subsequent risk of developing a PTSD, albeit only at lower methylation levels. In summary, the studies suggest that changes in expression of specific genes are associated with the experience of adversity and traumas at either early or later age, and as such, provide a potential molecular signature.
Implications for Practice and Research
There is increasing evidence for epigenetic involvement in stress- and trauma-related disorders, such as PTSD. Applying the concept of epigenetics to the field of PTSD represents an exciting frontier because of its ability to account for individual differences in response to trauma. Epigenetics might offer a biological framework for the biopsychological causes underlying PTSD by integrating both pre-existing risk factors and posttraumatic adaptations, and consequently help understand disease initiation and progression. Knowledge of the molecular mechanisms through which trauma and stressful experiences may alter the expression of genes critical to PTSD pathophysiology may also help foster the identification of etiologic subtypes of the disorder.
Recently, a new category of stress-associated disorder -called complex PTSD - has been proposed which takes into account exposure to prolonged stressors, usually involving multiple or repeated adverse events (Maercker et al., under review). Epigenetics could allow the validation of such proposed diagnostic entities by means of biologic correlates. Last but not least, reversible epigenetic changes that alter gene expression also represent attractive targets for therapeutic intervention. Currently, great expectations in the treatment of diseases are associated with the use of drugs that can modulate the activity of enzymes capable of causing epigenetic changes. Several “epigenetic drugs” have already been tested for the treatment of age-associated diseases (de Oliveira et al., 2012; Gryder et al., 2012; Price et al., 2012). Eventually, understanding the combined contribution of genetic and epigenetic factors to gene expression can lead to the development of a new array of prophylactic, diagnostic, and therapeutic applications.
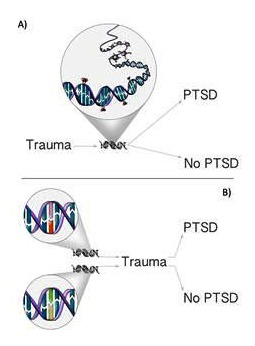
Illustrations of the two different ways of GxE interactions, both describing how genetic and environmental factors jointly influence the risk of developing a disease. A) Exposure to traumatic events (E) can lead to changes in DNA epigenetic profiles (such as methylation or chromatin structures) and as a consequence produce a specific phenotype. B) The effects of trauma exposure (E) differ according to an individual’s genetic predisposition (G) to develop a disease.
References
Binder, E.B., Bradley, R.G., Liu, W., et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of PTSD symptoms in adults. Journal of the American Medical Association, 299, 1291–1305.
Bradley, R.G., Binder, E.B., Epstein, M.P., et al. (2008). Influence of child abuse on adult depression: Moderation by the corticotropin-releasing hormone receptor gene. Archives of General Psychiatry, 65, 190–200.
de Oliveira, R. M., Sarkander, J., Kazantsev, A. G., and Outeiro, T. F. (2012). SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol, 3, 82.
Gryder, B. E., Sodji, Q. H., and Oyelere, A. K. (2012). Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 4, 505–524.
Higley, J.D., Hasert, M.F., Suomi, S.J., Linnoila, M. (1991). Nonhuman primate model of alcohol abuse: effects of early experience, personality and stress on alcohol consumption. Proc. Natl. Acad. Sci. USA, 18, 7261–7265.
Holliday, R. (1994). Epigenetics: An overview. Dev Genet, 15, 453-57
Russo, V.E.A., Martienssen, R.A., Riggs, A.D. (1996). Epigenetic Mechanisms of Gene Regulation. Monograph 22. Woodbury, NY: Cold Spring Harbor Laboratory Press.
Jaenisch, R., Bird, A. (2003). Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet, 22 (suppl), 245-54.
Klengel, T. Mehtal, D., Anacker, C., et al. (2012). Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nature Neuroscience. Epub ahead of print.
Koenen, K.C., Nugent, N.R., Amstadter, A.B. (2008). Gene-environment interaction in PTSD: Review, strategy and new directions for future research. European Archives on Psychiatry Clinical Neuroscience, 258(2), 82–96.
Koenen, K.C., Uddin, M., Chang, S.C., Aiello, A.E., Wildman, D.E., Goldmann, E., Galea, S. (2011). SLC6A4 methylation modifies the effect of the number of traumatic events on risk for posttraumatic stress disorder. Depression and Anxiety, 28, 639-647.
Maercker, A., Brewin, C.R., Bryant, R.A., et al. Proposals for mental disorders specifically associated with stress in the ICD-11. Lancet. Under review.
McGowan, P.O., Sasaki, A., D’Alessio, A.C., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience, 12(3), 342–348.
Meaney, M.J. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci, 24, 1161–1192.
Meaney, M.J., Szyf, M. (2007). Environmental programming of stress responses through DNA methylation: Life at the interface between a dynamic environment and a fixed genome. Dialogues in Clinical Neuroscience, 7, 103–123.
Price, N. L., Gomes, A. P., Ling, A. J., Duarte, F. V., Martin-Montalvo, A., North, B. J., et al. (2012). HDAC inhibitors for the treatment of cutaneous T-cell lymphomas. Future Med. Chem. 4, 471–486.
Renthal, W., Maze, I., Krishnan, V., et al. (2007). Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron, 56, 517–529.
Ressler, K.J., Mercer, K.B., Bradley, B., et al. (2011). Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature, 470, 492-497.
Szyf, M., Weaver, I.C., Champagne, F.A., Diorio, J. & Meaney, M.J. (2005). Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Frontiers in Neuroendocrinology, 26, 139–162.
Waddington, C. (1942). The epigenotype. Endeavour, 1, 18–20.
Weaver, I.C, Cervoni, N., Champagne, F.A., D'Alessio, A.C., Sharma, S., Seckl, J.R., Dymov, S., Szyf, M., Meaney, M.J. (2004). Epigenetic programming by maternal behavior. Nuroscience, 7(8), 847-54.
Yehuda, R., Bell, A., Bierer, L.M., Schmeidler, J. (2008). Maternal, not paternal, PTSD is related to increased risk for PTSD in offspring of Holocaust survivors. Journal of Psychiatric Research, 42, 1104-1111.
Andrea Burri is a doctoral-level clinical psychologist and genetic epidemiologist at the University of Zurich’s Department of Psychopathology and Clinical Intervention. She obtained her PhD degree at King’s College London, where she investigated the genetic epidemiology of female sexual dysfunction. The main focus of her current work is the identification of molecular mechanisms underlying stress-related and somatoform disorders.